Atualizado em 19 de maio de 2020 |
Publicado originalmente em 21 de maio de 2020
Conheça as principais características clínicas de cada subtipo de Atrofia Muscular Espinhal (AME), baseado na história natural da doença.
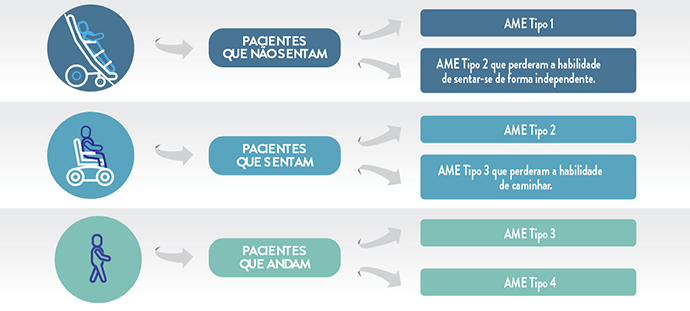
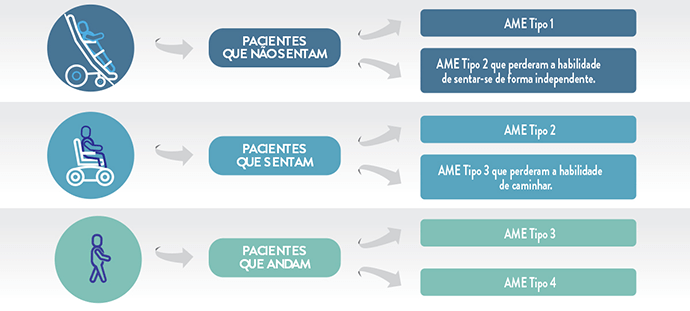
A Atrofia Muscular Espinhal (AME) é subdividida clinicamente em cinco tipos, definidos pela idade de aparecimento dos sintomas e pelas habilidades motoras alcançadas.
Assim, pessoas com a mesma doença podem apresentar diferentes níveis de acometimento, como indivíduos que não conseguem se sentar de forma independente, indivíduos que se sentam, mas não andam, ou indivíduos que andam, mas que podem perder essa habilidade com a progressão da doença.
Apesar das diferenças clínicas, pessoas com todos os tipos de AME têm a mesma doença, os sinais e sintomas são causados pela disfunção e morte de neurônios motores devido à diminuição da quantidade funcional de proteína SMN (1).
Conheça as principais características clínicas de cada subtipo de AME
É importante enfatizar que essas informações são baseadas na história natural da doença, de acordo com dados publicados em literatura científica ao longo dos últimos anos.
Sabemos que o advento e aprimoramento de cuidados multidisciplinares mais proativos, assim como o desenvolvimento de novas terapias, podem alterar de forma significativa os desfechos aqui listados (2).
As informações para cada subtipo de Atrofia Muscular Espinhal (AME) têm, portanto, caráter educativo, à medida que se considera a importância do entendimento completo sobre a história natural da doença para melhor definir as ações de cuidados integrais para a pessoa com AME.
Atrofia Muscular Espinhal (AME) - TIPO 0
É a forma mais grave de AME, e uma das mais raras. Tem início no período pré-natal e, além de acometimento motor e respiratório, pacientes com AME Tipo 0 podem apresentar alterações cardíacas e cerebrais (1,3).
Dificuldades respiratórias: Frequentemente necessitam de suporte ventilatório nos primeiros minutos ou horas após o nascimento.
Dificuldades motoras: hipotonia profunda, fraqueza grave e contraturas articulares.
Dificuldades de alimentação: apresentam grave disfagia e incapacidade de sucção para mamar.
Expectativa de vida: a grande maioria dos pacientes acaba falecendo nos primeiros dias ou semanas de vida, em geral não ultrapassando seis meses de idade.
Atrofia Muscular Espinhal (AME) - TIPO 1 Também conhecida como doença de Werdnig-Hoffman, é o subtipo mais comum de AME, correspondendo a cerca de 60% dos casos reportados em literatura. Sinais e sintomas têm início antes dos seis meses de vida (1,4,5).
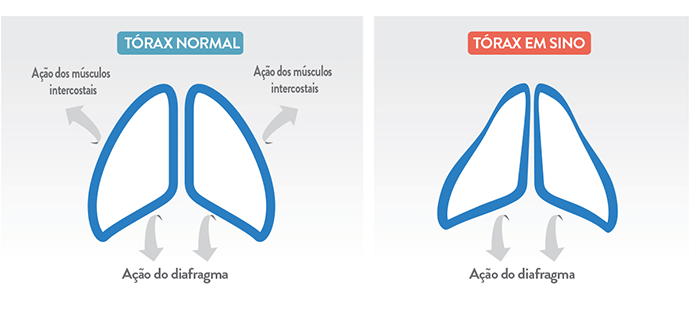
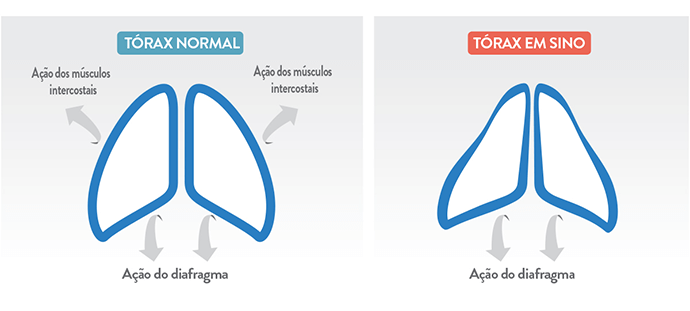
Dificuldades respiratórias: desenvolvem respiração paradoxal, e insuficiência respiratória é a principal causa de morbidade e mortalidade. A fraqueza e hipotonia da musculatura respiratória causam também deformidades no tórax, que assume um formato de sino.
Dificuldades motoras: não desenvolvem a capacidade de se sentar sem suporte e têm perda da maioria da movimentação ainda no primeiro ano de vida.
Dificuldades de alimentação: o acometimento de músculos da língua e faringe causa perda da capacidade de sucção ao mamar e disfagia, o que pode causar deficiência nutricional e risco de broncopneumonias de repetição. Crianças com AME tipo 1 podem necessitar de suporte nutricional via tubo gástrico. O acometimento da musculatura bulbar causa também fasciculações na língua, e pacientes com AME podem apresentar constipação intestinal.
Expectativa de vida: caso não sejam tomadas ações de tratamento, cerca de 68% dos pacientes morrem antes dos dois anos; e 84% antes dos quatro anos de idade. A adoção de cuidados respiratórios e nutricionais proativos pode reduzir a mortalidade antes dos 2 anos para 30%.
Atrofia Muscular Espinhal (AME) - TIPO 2
A AME 5q tipo 2, ou doença de Dubowitz, é a forma intermediária da doença em que os sintomas geralmente se iniciam entre os seis e dezoito meses de idade. Atinge cerca de 29% dos casos reportados na literatura (1,6,7).
Dificuldades respiratórias: os pacientes podem desenvolver hipoventilação, inicialmente durante o sono, requerendo o uso de suporte para respirar no período noturno, além de manobras para remoção de secreção.
Dificuldades motoras: pessoas com AME tipo 2 desenvolvem a capacidade de se sentar sem a necessidade de suporte, mas podem perder essa habilidade com a progressão da doença. Alguns pacientes conseguem ficar em pé, mas não conseguem andar de maneira independente. Muitos apresentam contraturas e deformidades articulares, incluindo escoliose grave.
Dificuldades de alimentação: com a progressão da doença, podem desenvolver disfagia e fraqueza na musculatura responsável pela mastigação e deglutição.
Expectativa de vida: estudos de história natural mostram que pacientes com AME tipo 2 chegam à idade adulta, no entanto, mostram mortalidade precoce em relação à população em geral.
Atrofia Muscular Espinhal (AME) - TIPO 3
Também conhecida como doença de Kugelberg-Welander, atinge cerca de 13% dos casos. Os primeiros sintomas aparecem após os dezoito meses de idade (1,6).
Dificuldades respiratórias: alguns pacientes desenvolvem dificuldade respiratória mais tardiamente, quando comparados ao tipo 2.
Dificuldades motoras: conseguem desenvolver a capacidade de andar independentemente, porém, em algum momento da vida, podem perder essa habilidade. Quanto mais precoce o início dos sintomas e sinais, mais cedo pode ocorrer a perda da marcha. As dificuldades ortopédicas, incluindo a escoliose, se agravam a partir do momento em que param de andar.
Dificuldades de alimentação: em casos mais graves ou com mais tempo de doença, podem desenvolver dificuldades para engolir.
Expectativa de vida: estudos mostram que a expectativa de vida destes pacientes pouco se diferencia da população não afetada.
Atrofia Muscular Espinhal (AME) - TIPO 4
Forma mais branda da doença, a AME tipo 4 é também uma das mais raras, representando menos de 5% dos novos casos. Na maioria das vezes, os primeiros sintomas aparecem a partir da segunda ou terceira década de vida. Pessoas com AME tipo 4 não apresentam dificuldades respiratórias ou de alimentação (1,9).
Dificuldades motoras: os pacientes podem apresentar hipotonia e reflexos musculares diminuídos, apresentando dificuldades, por exemplo, para subir e descer escadas ou para se levantar do chão. No entanto, levam a vida muito semelhante à população sem a doença.
Expectativa de vida: estes pacientes apresentam uma expectativa de vida semelhante à da população sem a doença.
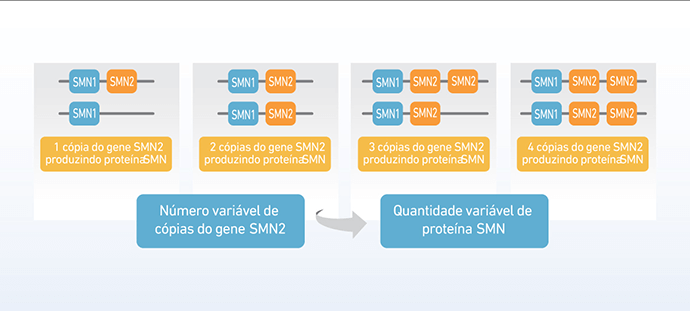
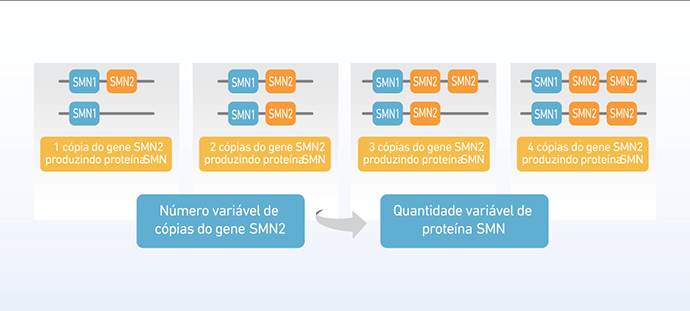
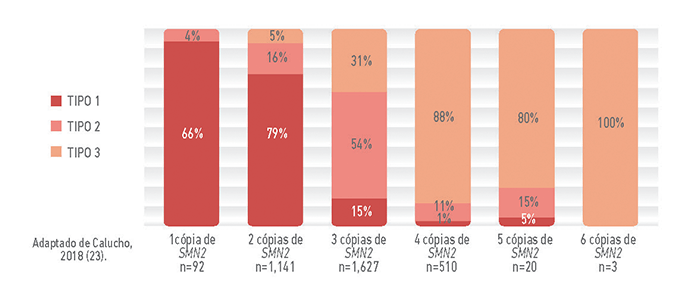
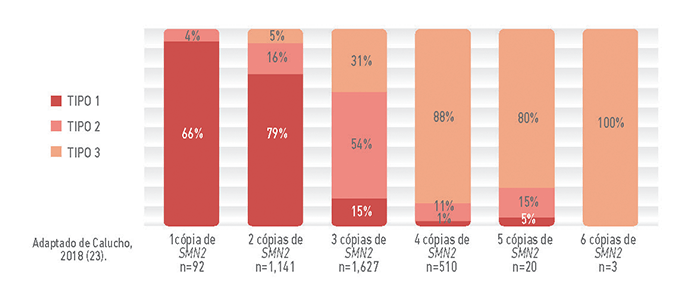
A correlação entre o número de cópias de SMN2 e o tipo de Atrofia Muscular Espinhal (AME)
Ao contrário da maioria dos genes, que geralmente estão presentes em duas cópias (uma vinda do pai e outra da mãe, o gene SMN2 pode ter número bastante variável de cópias. Assim, a quantidade de proteína SMN funcional produzida, a partir do gene SMN2, também será variável.
Nos pacientes com AME, que não produzem proteína SMN funcional a partir do gene SMN1, o gene SMN2 substitui parcialmente essa produção. Ainda assim, a quantidade de proteína funcional que o gene SMN2 produz não é suficiente para manter a sobrevivência dos neurônios motores e evitar a AME (9).
O gráfico abaixo mostra informações do tipo de AME e do número de cópias de SMN2 de aproximadamente 3500 pacientes. Observa-se que há uma correlação entre o número de cópias de SMN2 e o tipo de AME.
No entanto, essa correlação não é absoluta, assim, é importante destacar que o número de cópias de SMN2 não pode ser usado para determinar o prognóstico dos pacientes e nem, tampouco, pode ser usado para classificar o tipo de AME (9).
Referências Bibliográficas
1. Finkel R, Bertini E, Muntoni F, Mercuri E. 209th ENMC International Workshop: Outcome Measures and Clinical Trial Readiness in Spinal Muscular Atrophy 7-9 November 2014, Heemskerk, The Netherlands. Neuromuscul Disord. 2015;25(7):593–602.
2. Tizzano EF, Finkel RS. Spinal muscular atrophy: A changing phenotype beyond the clinical trials. Neuromuscul Disord. 2017;27(10):883–9.
3. Grotto S, Cuisset JM, Marret S, Drunat S, Faure P, Audebert-Bellanger S, et al. Type 0 Spinal Muscular Atrophy: Further Delineation of Prenatal and Postnatal Features in 16 Patients. J Neuromuscul Dis. 2016;3(4):487–95.
4. De Sanctis R, Pane M, Coratti G, Palermo C, Leone D, Pera MC, et al. Clinical phenotypes and trajectories of disease progression in type 1 spinal muscular atrophy. Neuromuscul Disord. 2018 Jan;28(1):24–8.
5. Darras BT. Spinal Muscular Atrophies. In: Pediatric Clinics of North America. Elsevier Inc; 2015. p. 743–66.
6. Zerres K, Rudnik-Schöneborn S, Forrest E, Lusakowska A, Borkowska J, Hausmanowa-Petrusewicz I. A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J Neurol Sci. 1997;146(1):67–72.
7. Faravelli I, Nizzardo M, Comi GP, Corti S. Spinal muscular atrophy—recent therapeutic advances for an old challenge. Nat Rev Neurol. 2015;11(6):351–9.
8. Zerres K, Rudnik-Schöneborn S. Natural history in proximal spinal muscular atrophy. Clinical analysis of 445 patients and suggestions for a modification of existing classifications. Neuromuscul Disord. 1995;52(5):518–23.
9. Calucho M, Bernal S, Alías L, March F, Venceslá A, Rodríguez-Álvarez FJ, et al. Correlation between SMA type and SMN2 copy number revisited: an analysis of 625 unrelated spanish patients and a compilation of 2,834 reported cases. Neuromuscul Disord. 2018;1–8.