Atualizado em 19 de maio de 2020 |
Publicado originalmente em 28 de março de 2020 às 13h18
Saiba mais sobre a Atrofia Muscular Espinhal (AME), uma doença rara, neuromuscular e genética que atinge um ou dois casos a cada 100 mil pessoas
A
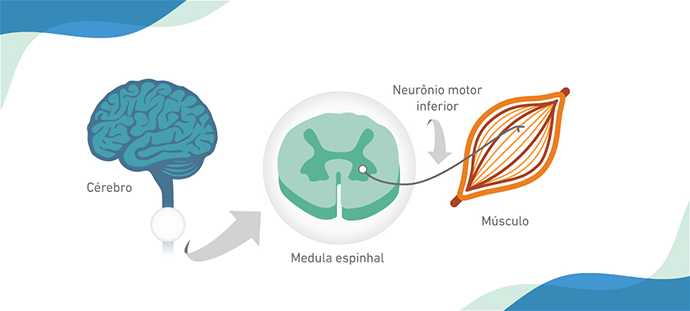
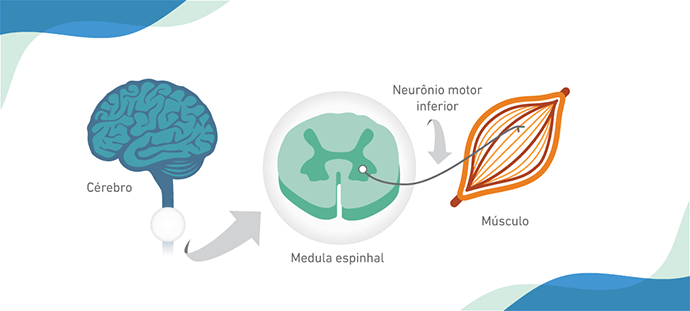
ções como sentar-se, andar, manter a cabeça ereta, se alimentar e respirar, entre outras, dependem do correto funcionamento dos músculos, que são controlados pelo cérebro e medula espinhal. Doenças que afetam os músculos e os neurônios que os controlam são conhecidas como doenças neuromusculares (1).
A Atrofia Muscular Espinhal (AME) é uma doença neuromuscular que causa a morte dos neurônios motores inferiores. Sem o comando desses neurônios, os músculos se degeneram e se tornam hipotônicos, fracos e atrofiam (2).
Pernas e membros mais próximos ao corpo geralmente são mais acometidos
É o comprometimento da função muscular que causa os sinais e sintomas da AME. Em geral, as pernas são mais acometidas que os braços, assim como as regiões dos membros mais próximos ao corpo em relação às extremidades (2).
É importante enfatizar que os neurônios responsáveis por sensações e dor não são comprometidos. Assim, ainda que pessoas com AME tenham dificuldades de movimentação, elas sentem toques, calor, frio e dor. Além disso, a AME não afeta neurônios do cérebro. Assim, pessoas com AME têm as funções cognitivas preservadas (2).
Causas da Atrofia Muscular Espinhal (AME): uma doença genética
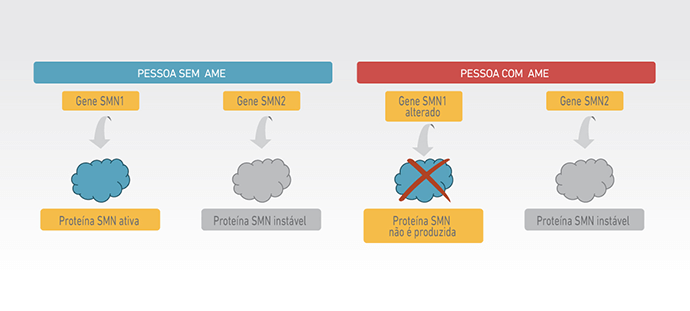
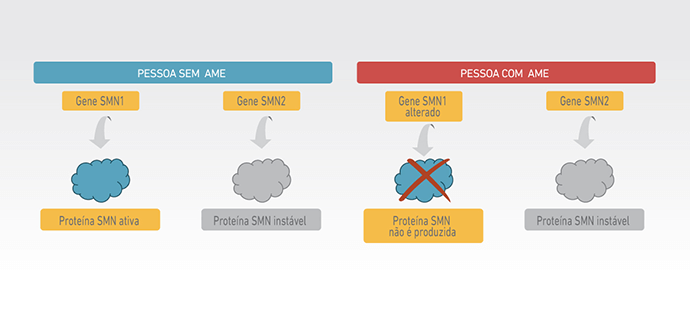
Na Atrofia Muscular Espinhal (AME) 5q, a disfunção, e posterior morte dos neurônios motores, é causada pela diminuição nos níveis da proteína de sobrevivência do neurônio motor, conhecida como proteína SMN (2).
A proteína SMN é produzida a partir do gene SMN1 que, em pacientes com AME, apresenta alterações genéticas que impedem sua função (3). A proteína SMN também pode ser produzida a partir de um segundo gene muito parecido com o SMN1, conhecido como gene SMN2. No entanto, somente pequena parte da proteína SMN produzida pelo gene SMN2 é funcional, não sendo suficiente para sustentar o funcionamento normal dos neurônios motores (2).
5 tipos de Atrofia Muscular Espinhal (AME)
A Atrofia Muscular Espinhal (AME) é subdividida clinicamente em cinco tipos, definidos pela idade de aparecimento dos sintomas e pelas habilidades motoras alcançadas. Assim, pessoas com a mesma doença podem apresentar diferentes níveis de acometimento, como indivíduos que não conseguem se sentar de forma independente, indivíduos que se sentam, mas não andam, ou indivíduos que andam, mas que podem perder essa habilidade com a progressão da doença (4).
Apesar das diferenças clínicas, pessoas com todos os tipos de AME têm a mesma doença, os sinais e sintomas são causados pela disfunção e morte de neurônios motores devido à diminuição da quantidade funcional de proteína SMN (4).
Atrofia Muscular Espinhal (AME) é uma doença hereditária
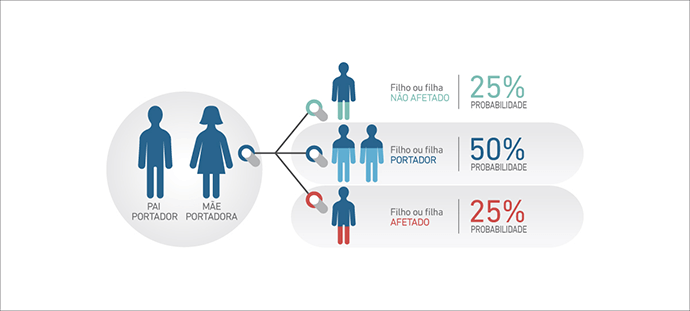
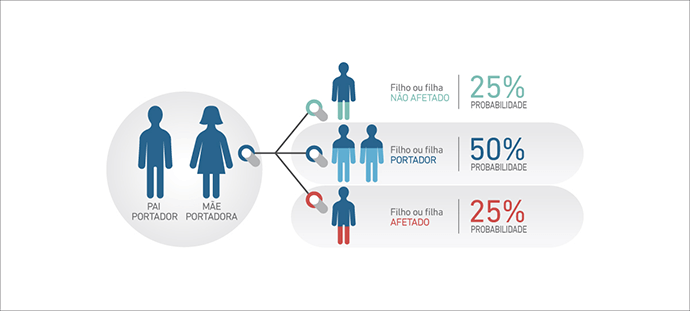
A Atrofia Muscular Espinhal (AME) 5q é uma doença genética de herança autossômica recessiva, o que significa que, para apresentar os sintomas da doença, os indivíduos devem possuir dois alelos SMN1 com alteração, um proveniente do pai e outro da mãe, na maioria dos casos (2).
O pai e a mãe, que possuem uma cópia do alelo alterado, são denominados portadores e não apresentam a doença. A chance que estes pais tenham um filho ou filha afetado é de 25% em cada gravidez.
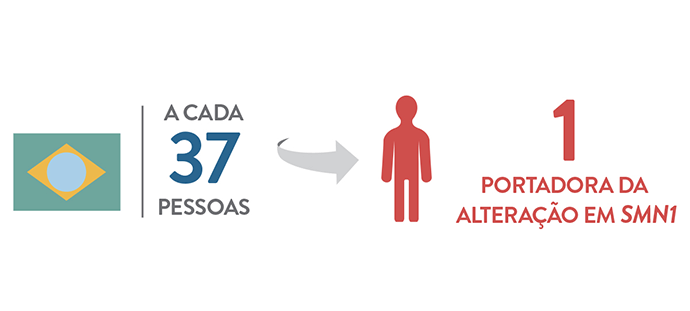
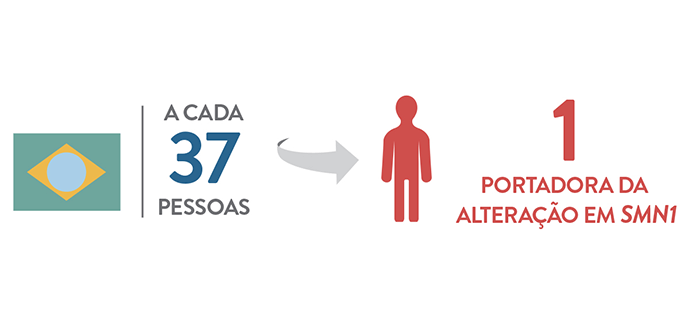
Estudos estimam que, em média, uma em cada 50 pessoas seja portadora do alelo SMN1 alterado, o que é uma frequência relativamente alta (5). Esse número varia de acordo com a população e no Brasil essa frequência é estimada em um portador a cada 37 pessoas (6).
Epidemiologia da Atrofia Muscular Espinhal (AME)
Não existem estudos de epidemiologia no Brasil, mas publicações internacionais mostram que a Atrofia Muscular Espinhal (AME) 5q tem incidência estimada em 1 caso a cada 10 mil nascidos vivos e prevalência de um a dois casos para cada 100 mil indivíduos (7).
Referências Bibliográficas
1. Kandel ER, Schwartz JH, Jessell TM, Siegelbaum SA, Hudspeth AJ. Principles of Neural Science. 5th ed. McGraw- Hill Medical; 2013. 1414 p.
2. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120–33.
3. Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, et al. Identification and characterization of a spinal muscular atrophy- determining gene. Cell. 1995;80(1):155–65.
4. Finkel R, Bertini E, Muntoni F, Mercuri E. 209th ENMC International Workshop: Outcome Measures and Clinical Trial Readiness in Spinal Muscular Atrophy 7-9 November 2014, Heemskerk, The Netherlands. Neuromuscul Disord. 2015;25(7):593–602.
5. Darras BT. Spinal Muscular Atrophies. In: Pediatric Clinics of North America. Elsevier Inc; 2015. p. 743–66.
6. Bueno KC, Gouvea SP, Genari AB, Funayama CA, Zanette DL, Silva WA, et al. Detection of spinal muscular atrophy carriers in a sample of the Brazilian population. Neuroepidemiology. 2011;36(2):105–7. Verhaart IEC, Robertson A, Wilson IJ, Aartsma-Rus A, Cameron S, Jones CC, et al. Prevalence, incidence and carrier frequency of 5q–linked spinal muscular atrophy – a literature review. Orphanet J Rare Dis. 2017;12(1):124.